引言
amber26 MD分子动力学模拟套件分为两个部分:ambertools26和amber26。ambertools包括了命令tleap,amber26包括了命令pmemd、pmemd.MPI、pmemd.cuda等命令。
amber26不能像很多软件一样直接提供.deb包或apt install 直接安装,而是从源码编译安装。从源码编译安装的过程,类似: https://blog.ztcdream.com/p/linux编译安装python
核心流程就是下载源码包,安装编译工具,配置cmake文件,编译,安装。而最麻烦、最容易报错的就是配置cmake文件和编译过程。
本教程在吃了好多报错之后😭,总结出一套适用于debian13的amber26编译安装方法。

下载源码包
amber26的官网是:Amber26官网
在下载页有两个文件可以下载,一个是Ambertools26,一个是Amber26。
需要tleap命令的就下载Ambertools26,需要pmemd命令就下载Amber26。
Ambertools26可以通过官网的提供的conda命令下载。对于Amber26,则必须通过源码编译安装。
与传统提供直链下载不同的是,下载页需要填写注册信息,名字和机构,才会获得有效的下载链接。实测信息可以随意填写。
源码tar包有300多M,而官网提供的链接不支持断点续传:一旦下载中途网络不稳定,就会终止下载,得到一个几KB的文本文件,要求重新填写信息,必须重新下载。
而国内访问官网下载,速度只有几十KB,很容易断连重下。使用cloudflare worker进行代理,速度很快但下载中途频繁断联,仍然要重新下载。因此需要很稳定的网络,或者用VPS(阿里云HK)帮忙下载。
为了获得下载链接,能够在VPS上下载,需要对网站抓包。
打开F12, 查看Network,勾选保留日志,刷新。在提交信息下载的时刻可查看到发起的post请求,尝试构造下载请求。
请求头
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
|
POST /cgi-bin/AmberTools26-get.pl HTTP/1.1
Accept: text/html,application/xhtml+xml,application/xml;q=0.9,image/avif,image/webp,image/apng,*/*;q=0.8,application/signed-exchange;v=b3;q=0.7
Accept-Encoding: gzip, deflate, br, zstd
Accept-Language: zh-CN,zh;q=0.9,en;q=0.8,en-GB;q=0.7,en-US;q=0.6,zh-TW;q=0.5
Cache-Control: max-age=0
Connection: keep-alive
Content-Length: 246
Content-Type: multipart/form-data; boundary=----WebKitFormBoundaryvToB6ELVIDd2cLp1
DNT: 1
Host: ambermd.org
Origin: https://ambermd.org
Referer: https://ambermd.org/GetAmber.php
Sec-Fetch-Dest: document
Sec-Fetch-Mode: navigate
Sec-Fetch-Site: same-origin
Sec-Fetch-User: ?1
Upgrade-Insecure-Requests: 1
User-Agent: Mozilla/5.0 (X11; Linux x86_64) AppleWebKit/537.36 (KHTML, like Gecko) Chrome/149.0.0.0 Safari/537.36 Edg/149.0.0.0
sec-ch-ua: "Microsoft Edge";v="149", "Chromium";v="149", "Not)A;Brand";v="24"
sec-ch-ua-mobile: ?0
sec-ch-ua-platform: "Linux"
|
表单
1
2
3
4
5
6
7
8
9
|
------WebKitFormBoundaryvToB6ELVIDd2cLp1
Content-Disposition: form-data; name="Name"
Zhang San
------WebKitFormBoundaryvToB6ELVIDd2cLp1
Content-Disposition: form-data; name="Institution"
USTC
------WebKitFormBoundaryvToB6ELVIDd2cLp1--
|
下载请求没有传入cookie,只是携带了姓名(Name)和机构(Institution)的两个表单项:

因此可以使用 wget 构造以下两个文件的下载链接:
Ambertools26:
1
2
3
4
5
6
7
|
wget -c \
--tries=0 \
--read-timeout=20 \
--post-data="Name=Ana+Fons&Institution=USTC" \
--header="Referer: https://ambermd.org/GetAmber.php" \
-O AmberTools26-get.pl \
"https://ambermd.org/cgi-bin/AmberTools26-get.pl"
|
Amber26:
1
2
3
4
5
6
7
|
wget -c \
--tries=0 \
--read-timeout=20 \
--post-data="Name=Zhang+San&Institution=USTC" \
--header="Referer: https://ambermd.org/GetAmber.php" \
-O Amber26free-get.pl \
"https://ambermd.org/cgi-bin/Amber26free-get.pl"
|
有了wget命令,就可以在网络良好的VPS上下载,然后把文件传回本地待部署amber的服务器上。
文件体积较大,经过了几次转手,最终上传到服务器后,必须对文件进行校验,检测文件在网络传输过程是否完整。
-
AmberTools26
改名AmberTools26-get.pl 为 AmberTools26.tar.bz2:
1
|
mv AmberTools26-get.pl AmberTools26.tar.bz2
|
验证 md5sum 是否为一致(370620f5ddb646e552a182ce8625b78e.):
1
|
md5sum AmberTools26.tar.bz2
|
-
Amber26
同样的重命名文件为pmemd26.tar.bz2:
1
|
mv Amber26free-get.pl pmemd26.tar.bz2
|
检查 md5sum 是否一致(ceeabc133e772c115183d5cf6a87676b):
到这一步为止,你将会获得两个文件:AmberTools26.tar.bz2,pmemd26.tar.bz2。并且已经验证了文件的完整性,并将其上传到待安装的服务器上。
Amber26(单核版)编译安装
Amber套件分成两个部分,Ambertools的安装不必从源码编译,可直接使用conda安装。因此接下来只重点介绍amber26的源码编译与安装。
注意:默认情况下,原始编译配置文件的amber26不支持cuda加速(pmemd.cuda),也不支持多核cpu加速(pmemd.MPI)。为了教程的通用性,减少因为硬件和系统差异造成的编译失败,接下来安装的是默认版本(pmemd),加速优化会在后文提及。
解压源码包
在服务器上创建一个专门的文件夹,如 amberDir,amber的编译以及生成的可执行文件将会放在这里。
将pmemd26.tar.bz2移动到amberDir,解压:
1
|
tar -xjvf pmemd26.tar.bz2
|

解压后获得文件夹 pmemd26_src,里面包含了编译所需的源码。
官网的下载页面给出的文件名是 pmemd26.tar.bz2,解压后文件夹为pmemd26_src,但在安装页面的教程使用的目录是amber26_src,前后不一致(历史遗留问题)。本文中仍然使用pmemd26_src。
编译
cd pmemd26_src/build,准备进行编译。
在编译之前,需要安装相应的编译工具:
以debian-13为例,官网推荐的:
1
2
3
4
5
6
|
apt -y install python3 tcsh make \
gcc gfortran g++ \
flex bison patch bc \
libbz2-dev libzip-dev \
xorg-dev wget \
rsync openssh-client
|
还要安装cmake:
全部安装之后,使用pwd确认工作目录处于 pmemd26_src/build文件夹,再使用命令进行编译。
提示:在性能较弱的电脑上,编译amber26会很久。为了避免编译中途ssh连接断开,任务中断,在编译之前请使用screen或tmux创建一个虚拟会话。
运行 run_cmake:
编译并安装:
在编译过程中,会输出许多 Warning,这是正常的,重点看最后几行输出是否正常。
1
2
3
4
5
6
7
8
9
10
11
|
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/mbar/win_0.30.dumpave
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/mbar/win_0.00.dumpave
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/mbar.chk.save
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/wtp.metafile
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/wtp.out.save
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/wtp.chk.save
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/wtp
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/wtp/win_0.20.dumpave
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/wtp/win_0.10.dumpave
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/wtp/win_0.30.dumpave
-- Installing: /home/ztc/amberDir/pmemd26/test//./ndfes/wtp/win_0.00.dumpave
|

最后输出了许多 -- Installing ,没有报错,查看$?为0,说明编译过程无异常。
编译之后,amberDir目录下会产出一个 pmemd26 文件夹(之前的是pmemd26_src)。该文件夹下的bin里即为pmemd26的可执行文件。


添加环境变量
接下来就是把该文件夹添加到环境变量中:
将下面的代码中的/home/ztc/amberDir/pmemd26前面的用户路径修改为实际的路径,并添加到 .bashrc中。
1
2
|
export AMBERHOME=/home/ztc/amberDir/pmemd26
source $AMBERHOME/amber.sh
|
使用nano ~/.bashrc 或 vim ~/.bashrc 编辑.bashrc,把上面修改后的两行添加到文件末尾,保存退出。
执行 source ~/.bashrc使其生效。
运行test脚本
安装好的amber目录下自带了一个测试的文件,可以测试amber是否能正常使用。
1
2
3
|
cd /home/ztc/amberDir/pmemd26
source ~/.bashrc
make test.serial
|
如果出现明显的报错,检查一下环境变量。运行结束后,正常的输出应当是这样的:
1
2
3
4
5
6
7
8
9
|
Finished serial test suite for Amber at 2026年 06月 07日 星期日 12:20:55 CST.
make[2]: 离开目录“/home/ztc/amberDir/pmemd26/test”
214 file comparisons passed
4 file comparisons failed (4 of which can be ignored)
0 tests experienced errors
Test log file saved as /home/ztc/amberDir/pmemd26/logs/test_amber_serial/2026-06-07_12-18-36.log
Test diffs file saved as /home/ztc/amberDir/pmemd26/logs/test_amber_serial/2026-06-07_12-18-36.diff
make[1]: 离开目录“/home/ztc/amberDir/pmemd26/test”
|
214个文件通过了测试,4个文件的错误可以被忽略,0个意外的错误。
到这一步,成功编译了amber26的默认版本,并添加到环境变量。可以使用pmemd命令了。
Amber26(多核版)安装
刚才安装得到的pmemd,是pmemd单核CPU版本,不能充分调动CPU,基本没法用的。
源码包默认的run_cmake文件,默认配置没有开启多核,也没有启用CUDA。
run_cmake核心的配置是这一段(Linux):
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
# Assume this is Linux:
cmake $AMBER_PREFIX/pmemd26_src -Wno-dev \
-DCMAKE_INSTALL_PREFIX=$AMBER_PREFIX/pmemd26 \
-DCOMPILER=GNU \
-DMPI=FALSE -DCUDA=FALSE -DINSTALL_TESTS=TRUE \
-DDOWNLOAD_MINICONDA=FALSE -DBUILD_PYTHON=FALSE \
-DBUILD_PERL=FALSE -DBUILD_GUI=FALSE \
-DPMEMD_ONLY=TRUE -DCHECK_UPDATES=FALSE \
-DFORCE_EXTERNAL_LIBS=mkl -DMKL_MULTI_THREADED=FALSE \
-DPMEMD_XRAY_CPU_FFT_BACKEND=MKL \
2>&1 | tee cmake.log
fi
|
-DMPI=FALSE:多核版本-DCUDA=FALSE: CUDA版本
现在都是未启用。
要编译多核版本,-DMPI=FALSE改为-DMPI=TRUE。
此外,还要安装调度多核的工具:
1
|
sudo apt install openmpi-bin libopenmpi-dev
|
然后加上一行:
1
|
-DMPI_C_COMPILER=/usr/bin/mpicc \
|
并关闭python检测:
或者直接复制替换:
1
2
3
4
5
6
7
8
9
10
11
12
13
|
cmake $AMBER_PREFIX/pmemd26_src -Wno-dev \
-DCMAKE_INSTALL_PREFIX=$AMBER_PREFIX/pmemd26 \
-DCOMPILER=GNU \
-DMPI_C_COMPILER=/usr/bin/mpicc \
-DMPI=TRUE \
-DCUDA=FALSE \
-DPGM=FALSE \
-DINSTALL_TESTS=TRUE \
-DDOWNLOAD_MINICONDA=FALSE \
-DBUILD_PYTHON=FALSE \
-DPMEMD_ONLY=TRUE \
-DCHECK_UPDATES=FALSE
2>&1 | tee cmake.log
|

1
2
3
|
./clean_build
./run_cmake
make install
|
不出意外,应该编译成功:

ambertools26不建议随pmemd编译安装,实测成功率较低,会卡在python环境和它指定的miniConda的问题。这里就遵循官网的建议单独使用conda安装ambertools26。
官网的下载页给出了很简单的命令,实际运行下来并不能顺利的安装,会出现conda包太大下载超时的问题,下面的教程会着重解决这个问题。
安装conda
使用conda安装ambertools26之前,当然得装conda。conda可以通过现成的脚本一键安装:
1
|
wget https://github.com/conda-forge/miniforge/releases/latest/download/Miniforge3-Linux-x86_64.sh
|
下载好安装脚本,用chomd +x赋权。
注意:接下来运行脚本时,建议使用普通用户(或当前用户),这样conda会装到用户所在的目录。
运行脚本:
1
|
bash Miniforge3-Linux-x86_64.sh
|
一路回车,使用默认选项即可。
在最后这里,选择yes(默认是no):
1
2
3
4
5
6
7
8
|
Do you wish to update your shell profile to automatically initialize conda?
This will activate conda on startup and change the command prompt when activated.
If you'd prefer that conda's base environment not be activated on startup,
run the following command when conda is activated:
conda config --set auto_activate_base false
Note: You can undo this later by running `conda init --reverse $SHELL`
Proceed with initialization? [yes|no]
[no] >>>
|
选择 yes 可以让脚本自动帮你把 Conda 的路径和初始化代码写入到你的 ~/.bashrc 文件中。
安装成功后,终端提示符前会显示 (base),这是激活了conda环境。
用conda给ambertools创建一个虚拟环境:
1
|
conda create --name AmberTools26 python=3.12
|
激活刚才创建的环境:
1
|
conda activate AmberTools26
|
终端提示符应当变成 (AmberTools26)。
配置conda镜像站
添加北外镜像的 conda-forge 换源:
1
|
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/conda-forge/
|
保持 strict 优先级,Conda 会优先去国内镜像匹配:
1
|
conda config --set channel_priority strict
|
将连接超时和读取超时时间改大(比如改成 60 秒):
1
2
|
conda config --set remote_connect_timeout_secs 60.0
conda config --set remote_read_timeout_secs 60.0
|
离线安装
接下来很关键了,如果直接运行官网的conda install dacase::ambertools-dac=26,conda将从网络源中下载所有要用的包。即便配置了国内的镜像站,仍然会有一个900多MB的.conda包下载超时。
这个.conda包要单独用浏览器下载,传到服务器上。下面是用wget:
1
|
wget "https://conda.anaconda.org/dacase/linux-64/ambertools-dac-26.0.0-py312h2009f2f_0.conda"
|
下载之后用conda离线安装:
1
|
conda install ./ambertools-dac-26.0.0-py312h2009f2f_0.conda
|
再用conda install dacase::ambertools-dac=26在线安装剩下的包:
1
|
conda install --only-deps dacase::ambertools-dac=26 -y
|
--only-deps:只安装剩余的包。
这次就能顺利安装了。
使用
激活环境:
1
|
conda activate AmberTools26
|
挂载 Conda 环境下的 Amber 路径:
1
|
source $CONDA_PREFIX/amber.sh
|
现在就可以使用 AmberTools 的工具了,比如:
Amber26(cuda版)编译
安装cuda.run
在cuda官网: (https://developer.nvidia.com/cuda-downloads?target_os=Linux&target_arch=x86_64
下载对应cuda文件,选择runfile文件。

运行安装程序,在安装界面accept用户协议,用空格勾选安装cuda-toolkit,空格取消安装driver。

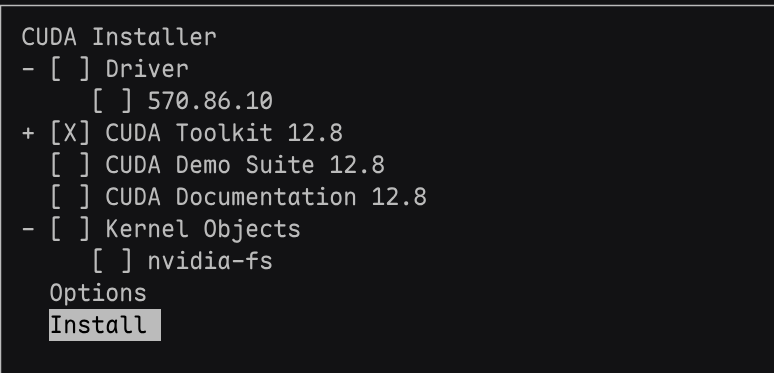
安装之后的cuda位于/usr/local:

mamba配置虚拟环境
像之前直接使用系统的gcc编译工具,很难成功编译cuda版。根据知乎的方法,用mamba来创建一个虚拟环境专门给amber编译用。
原文链接:https://zhuanlan.zhihu.com/p/2039652029346402710
1
2
3
4
|
mamba env create -n amber26 gcc=12 gxx=12 gfortran=12 -y
mamba activate amber26
mamba install zlib -y
mamba install mpich -y
|
把之前的run_cmake对应内容改成:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
-DCOMPILER=GNU \
-DSTATIC=OFF \
-DMPI=OFF \
-DCUDA=ON \
-DINSTALL_TESTS=OFF \
-DDOWNLOAD_MINICONDA=OFF \
-DBUILD_PYTHON=OFF \
-DBUILD_PERL=OFF \
-DBUILD_GUI=OFF \
-DPMEMD_ONLY=ON \
-DCHECK_UPDATES=OFF \
-DCMAKE_SHARED_LINKER_FLAGS="-lrt -lpthread -ldl" \
-DCMAKE_EXE_LINKER_FLAGS="-lrt -lpthread -ldl" \
-DCUDA_TOOLKIT_ROOT_DIR=/usr/local/cuda-12.8 \
-DCMAKE_LIBRARY_PATH=$CONDA_PREFIX/x86_64-conda-linux-gnu/sysroot/usr/lib
|
之后:./run_cmake,看看也没有报错。
没有报错,在继续make install之前,screen开一个虚拟终端,cuda版我编译了一个小时。
没问题的话,一个小时后pmemd.cuda就编译出来了。